Transforming Lives through Innovative Cancer Science
In this section, you will learn:
- Research that increases our understanding of the genetic, molecular, and cellular characteristics of cancer is continuing to spur advances in the treatment of cancer.
- Advances are being made across all five pillars of cancer care: surgery, radiation, cytotoxic chemotherapy, molecularly targeted therapy, and immunotherapy.
- From August 1, 2018 to July 31, 2019, the FDA approved 17 new therapeutics for treating patients with certain types of cancer.
- During the same period, the uses of 10 previously approved anticancer therapeutics were expanded by the FDA to include the treatment of additional types of cancer.
Contents
Contents
Progress across the continuum of clinical cancer care improves survival and quality of life for people around the world. The progress is driven by the dedicated efforts of individuals working throughout the cycle of medical research (see Figure 7).
Medical Research

Medical research is an iterative cycle (see Figure 7). Each discovery builds on knowledge gained from prior research. The cycle is set in motion when discoveries with the potential to affect the practice of medicine and public health are made in any area of medical research or clinical practice (see sidebar on What Is Medical Research?). The discoveries lead to hypotheses that are tested by researchers performing experiments in a wide range of models that mimic healthy and diseased conditions. Results from these experiments can lead to the identification of a potential preventive intervention or therapeutic target, or to the identification of a potential predictive or prognostic biomarker. They also can feed back into the cycle by providing new discoveries that lead to more hypotheses.
After a potential therapeutic target is identified, it takes many more years of preclinical research before a candidate therapeutic is developed and ready for testing in clinical trials (see sidebar on Therapeutic Development).
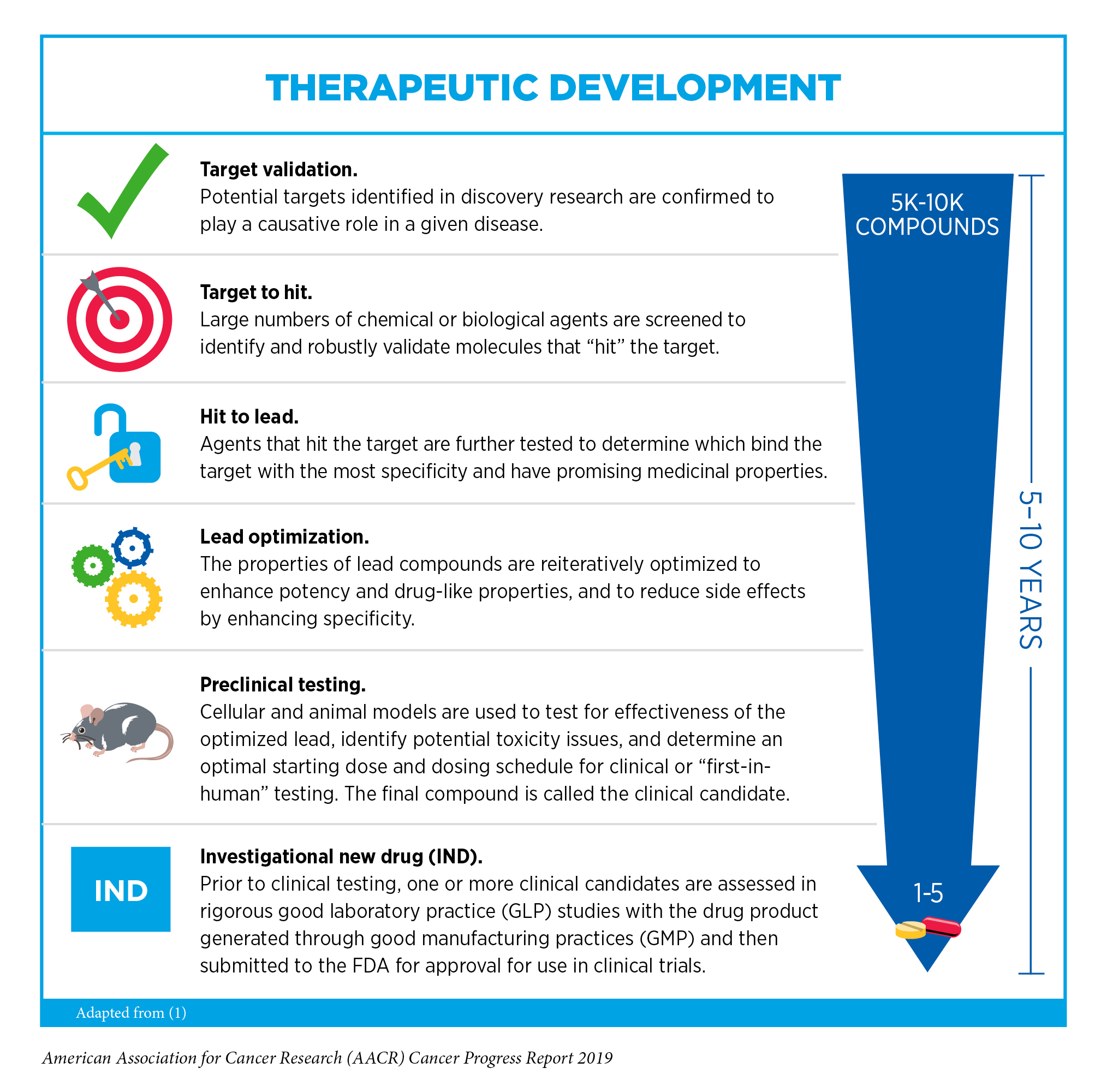
During this time, candidates are rigorously tested to identify potential toxicities and to determine the appropriate doses and dosing schedules for testing in the first clinical trial.
Before candidate therapeutics can be approved by the FDA and used as part of patient care, the safety and efficacy of the agents must be rigorously tested through clinical trials. All clinical trials are reviewed and approved by institutional review boards before they can begin and are monitored throughout their duration. There are several types of cancer clinical trials, including treatment trials, prevention trials, screening trials, and supportive or palliative care trials, each designed to answer different research questions.

Clinical trials that test candidate therapeutics for patients with cancer have traditionally been done in three successive phases (see Figure 8). However, the multiphase clinical testing process requires a large number of patients and takes many years to complete, making it extremely costly and one of the barriers to rapid translation of scientific knowledge into clinical advances. One recent report estimated that the average time from patent filing through clinical development and FDA approval to market launch was 13.6 years for the 59 new therapeutics approved by the FDA in 2018, of which 16 were new therapeutics for patients with cancer (161). Another study estimated that the median time it takes to complete the multiphase clinical testing process for anticancer therapeutics is 13.1 years (162).
Over the past three decades, the FDA implemented several changes that have altered how clinical trials can be conducted and reviewed in an effort to reduce the length of time it takes to obtain a clear result from a clinical trial, including developing four evidence-based strategies to expedite assessment of therapeutics for life-threatening diseases such as cancer. Two of these strategies, accelerated approval and breakthrough therapy designation, were recently shown to be working as intended: The average time from patent filing to launch for therapeutics approved using the accelerated approval and breakthrough therapy designation strategies was 15 percent and 19 percent shorter, respectively, than it was for therapeutics approved without using these strategies (161). This progress in FDA assessment of new therapeutics is highly relevant to patients with cancer because most anticancer therapeutics are approved using one or more of the four expedited strategies. For example, of the 17 new anticancer therapeutics approved by the FDA during the 12 months spanning this report, 16 were approved using one of the four expedited review strategies, including nine that were approved using the accelerated approval and/or breakthrough therapy designation strategies.
In addition, advances in our understanding of cancer biology have enabled researchers, regulators, and representatives of the pharmaceutical industry to develop new ways of designing and conducting clinical trials. Among the new ways to design clinical trials that have emerged in recent years are adaptive, seamless, and master protocol designs (163-165). These designs aim to streamline the clinical development of new anticancer therapeutics by matching the right therapeutics with the right patients earlier, reducing the number of patients who need to be enrolled in the trial before it is determined whether the anticancer therapeutic being evaluated is safe and effective, and/or decreasing the length of time it takes for a new anticancer therapeutic to be tested and made available to patients if the trial shows it is safe and effective.
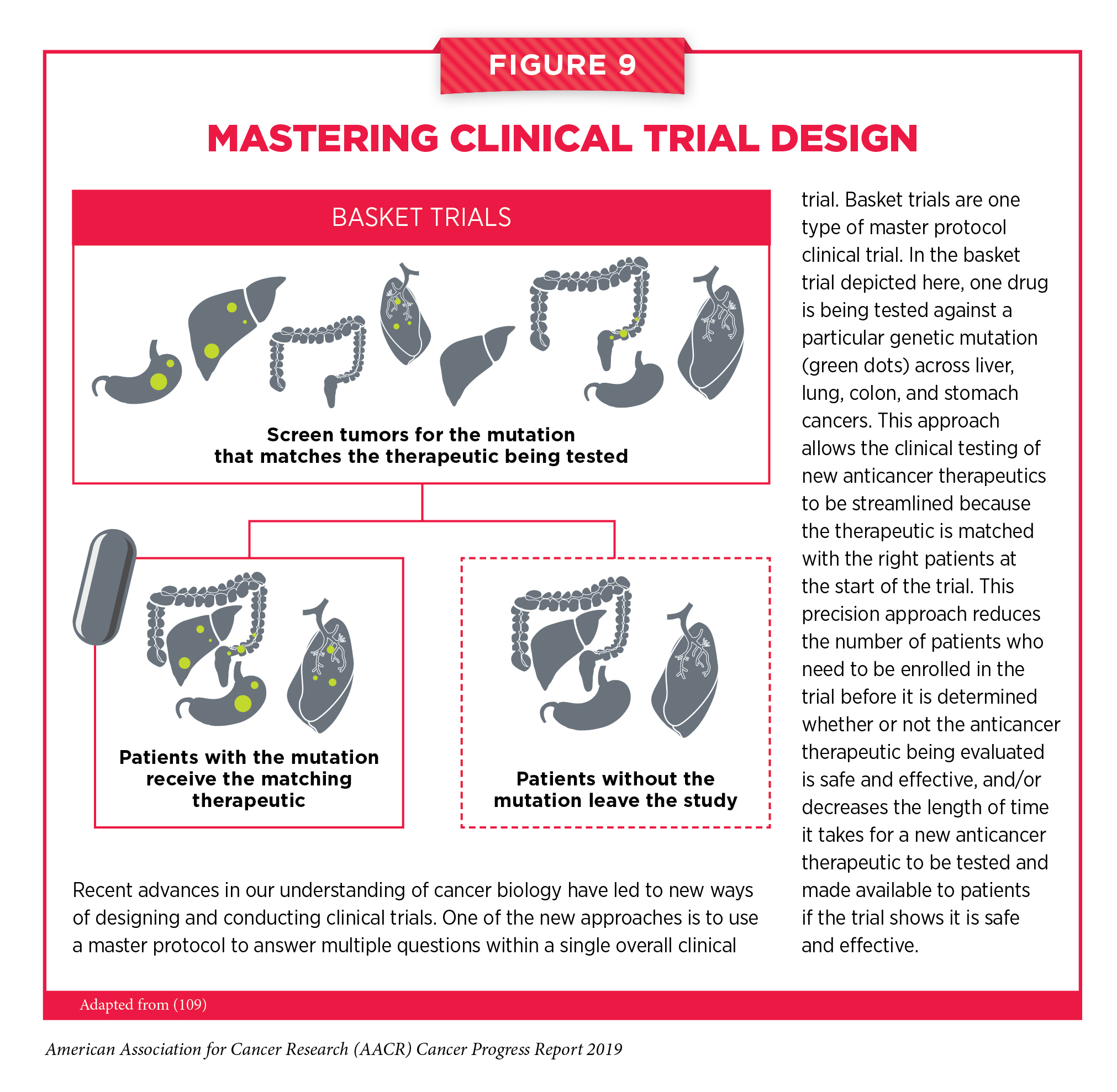
Master protocol design clinical trials aim to answer multiple questions within a single overall clinical trial (165). The emergence of this clinical trial design has largely been driven by our increased understanding of the genetic mutations that underpin cancer initiation and growth. “Basket trials” are one example of genetic mutation – based master protocol design clinical trials (see Figure 9). These trials allow researchers to test one anticancer therapeutic on a group of patients who all have the same type of genetic mutation, regardless of the anatomic site of the original cancer. One new molecularly targeted therapeutic that was shown to work against an array of cancer types characterized by a specific genetic feature, or biomarker, in a number of basket trials is highlighted in Targeting Cancers Based on Tumor Biomarker, Not Tumor Origin (166).

Two recent reports show that using biomarkers, such as the presence of a specific genetic mutation, does help to increase the efficiency of the clinical development of new therapeutics (162, 167). One report found that when considering all areas of medicine, candidate therapeutics entering phase I clinical trials that were matched to patients using biomarkers had a 25.9 percent chance of FDA approval, compared with 8.4 percent for candidates that were not matched using biomarkers (167). Another study looking at anticancer therapeutics estimated that the chance of FDA approval was 10.7 percent for candidate agents that were matched to patients using biomarkers, compared with 1.6 percent for unmatched candidates (162). These data indicate that we need to do more to improve the clinical trial enterprise.
Other pressing challenges that need to be overcome are low participation in clinical trials, in particular among adolescents and young adults, and a lack of diversity among those who do participate (168-171) (see sidebar on Disparities in Cancer Clinical Trial Participation). Low participation in clinical trials means that some trials fail to enroll enough participants to draw valid conclusions about the effectiveness of the medical product being tested (172-173). Understanding and then overcoming barriers to clinical trial participation for all segments of the population is vital if we are to accelerate the pace of progress against cancer for all.
Progress across the Clinical Cancer Care Continuum
Research discoveries made as a result of innovative cancer science are continually being translated to new medical products for cancer prevention, detection, diagnosis, treatment, and survivorship. The approval of new medical products is not the end of a linear research process. Rather, it is an integral part of the medical research cycle because observations made during the routine use of new medical products can be used to accelerate the pace at which similar products are developed and to stimulate the development of new, more effective products.

In addition, observations made during the real-world use of a product can be utilized to further enhance the use of that product. For example, the FDA utilized data from the real-world use of a molecularly targeted therapeutic called palbociclib (Ibrance) to approve the use of palbociclib as a treatment for men with advanced or metastatic breast cancer that tests positive for hormone receptor (HR) and negative for HER2 in April 2019. Given that there are only 2,670 men expected to be diagnosed with breast cancer in 2019 in the United States, conducting rigorous clinical trials of new treatments for these individuals is challenging. Thus, the use of real-world data to support FDA decision-making has accelerated the pace of progress for men with breast cancer such as Kirby Lewis.

New FDA-approved medical products are usually utilized alongside treatments already in use, including surgery, radiotherapy, and cytotoxic chemotherapy, which continue to be the mainstays of clinical cancer care (see Figure 10) (see
Supplemental Table 2a, 2b, 2c, 2d, and Supplemental Table 3a,
3b).

The following discussion focuses primarily on the 17 new anticancer therapeutics approved by the FDA in the 12 months spanning this report, August 1, 2018 to July 31, 2019 (see Table 5). Also highlighted are the 10 previously approved anticancer therapeutics that received FDA approval for treating additional types of cancer in that period. Not discussed are FDA approvals related to expanding the use of an anticancer therapeutic previously approved for a given type of cancer to include treatment with that therapeutic at different timepoints during the treatment of the same cancer type. For example, the May 2019 FDA approval expanding the use of the molecularly targeted therapeutic ado-trastuzumab emtansine (Kadcyla) to include postsurgery, or adjuvant, treatment of women with early-stage HER2-positive breast cancer (see Figure 11). This expansion, which occurred more than 6 years after the molecularly targeted therapeutic was first approved for treating metastatic HER2-positive breast cancer, was based on results from a phase III clinical trial that showed that adjuvant ado-trastuzumab emtansine treatment reduced the risk of disease recurrence by half compared with standard treatment (176).
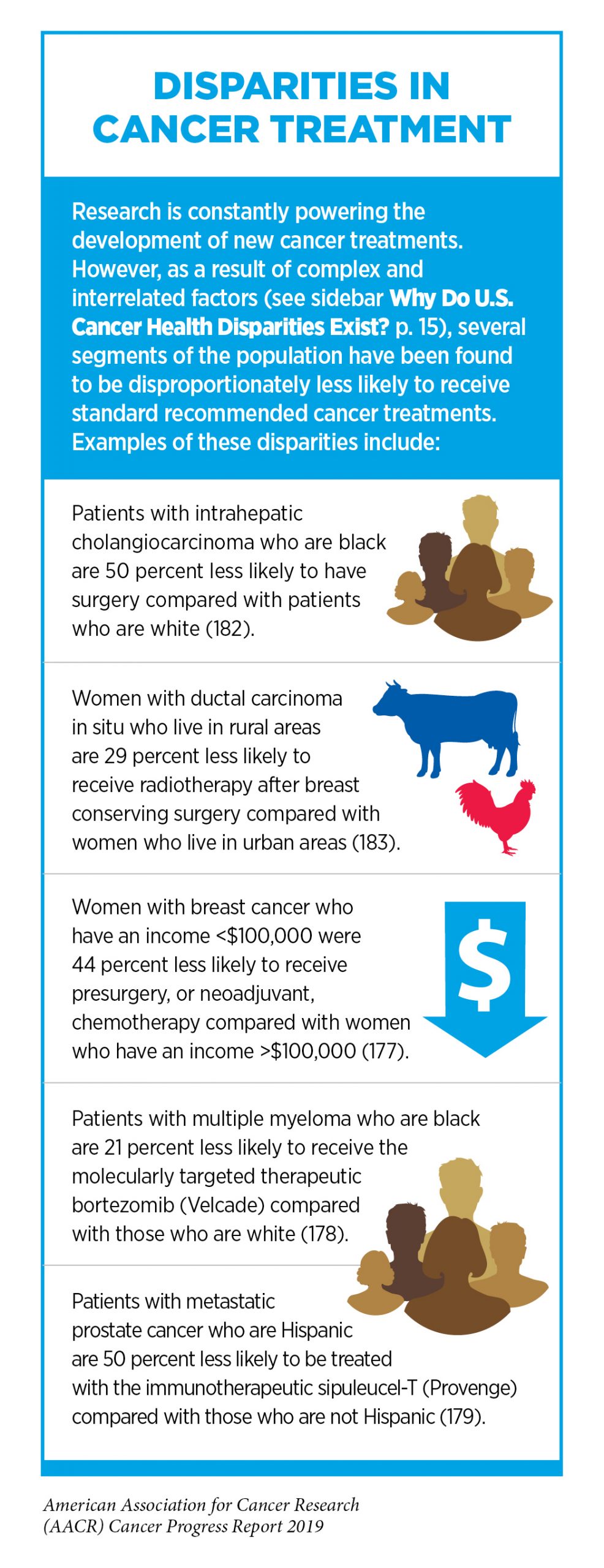
New medical products used across the continuum of clinical cancer care transform lives by improving survival and quality of life. However, not all patients receive the standard of care recommended for the type and stage of cancer that they have been diagnosed with (177-179) (see sidebar on Disparities in Cancer Treatment). Thus, it is imperative that all stakeholders committed to driving progress against cancer work together to address the challenge of disparities in cancer treatment because these can be associated with adverse differences in survival. In fact, two recent studies showed that disparities in multiple myeloma and prostate cancer survival for African Americans compared with whites were eliminated if they had equivalent access to care and to standard treatments (180-181).
Treatment with Surgery
For many years, surgery was the only pillar of cancer treatment (see Figure 10). Today, it remains the foundation of curative treatment for many patients (184). One study found that patients diagnosed at the earliest stage, stage 1, were more than five times as likely to be treated with surgery as patients diagnosed at the most advanced stage, stage 4 (185). Given the curative potential of surgery, these data highlight the important role of diagnosing cancer at the earliest possible stage.
Despite the immense benefits of surgery, complications are common and can negatively affect patient quality of life (186-187). Enhanced recovery after surgery (ERAS) programs are emerging as one approach to address this issue. These multimodal, transdisciplinary programs focus on optimizing preoperative, intraoperative, and postoperative patient care using strategies that ensure the patient is as physically and emotionally fit for surgery as possible, alleviate the stress of surgery, promote recovery, and reduce the time before patients can begin adjuvant treatment. Providing patients with an individualized prehabilitation plan that includes exercise, nutrition, stress reduction, and smoking cessation components to optimize their physical fitness before surgery is one preoperative strategy included in some ERAS programs (188-190). The components of ERAS programs can vary depending on the type of surgery being performed and the center at which the surgery is being performed, but overall these programs have been successful. One study found that among patients undergoing surgery for lung cancer, the rate of postsurgery complications was significantly lower for those who participated in ERAS programs than it was for those who did not participate (187). Another showed that among patients undergoing surgery for colorectal cancer, those who participated in a prehabilitation plan that included exercise, protein supplementation, and relaxation were significantly more likely to rapidly regain their presurgery functional walking capacity than those who did not participate (191, 189).
Another approach to reducing the complications and improving quality of life after surgery is to perform less invasive surgery. Before such approaches to surgery can become standard of care, it is important that they are shown in rigorous, well-designed, large clinical trials to have no adverse effect on patient survival. The importance of such trials is highlighted by several recent studies showing that less invasive surgery may only benefit patients with certain types of cancer. One clinical trial showed that a less invasive form of surgery for esophageal cancer called hybrid minimally invasive esophagectomy resulted in fewer major complications during and after surgery than open esophagectomy, and did not compromise disease-free and overall survival (192). In contrast, two other studies showed that minimally invasive surgery for early-stage cervical cancer is associated with shorter overall survival compared with open surgery (193, 194).
Reducing complications and improving quality of life after surgery can also be achieved by performing less extensive surgery. Two clinical trials recently found that surgical removal of large numbers of lymph nodes in the area around a cancer does not benefit all patients, and can be a source of surgical complications and long-term adverse effects (195, 196). In one trial, women who had early-stage breast cancer with defined clinical characteristics had equally good disease-free survival after 10 years whether or not they had an invasive surgical procedure called axillary lymph node dissection during or after breast cancer surgery. In the other trial, women with advanced ovarian cancer had equally good overall survival regardless of whether they had an invasive surgical procedure called a systematic pelvic and paraaortic lymphadenectomy as part of ovarian cancer surgery. In both trials, the women who did not have the additional surgical procedure had fewer complications after surgery.
Treatment with Radiotherapy

Radiotherapy became the second pillar of cancer treatment in 1896 (see Figure 10). Today, about 50 percent of patients receive radiotherapy to shrink or eliminate tumors or to prevent local recurrence (184) (see sidebar on Using Radiation in Cancer Care).
Despite the immense benefits of radiotherapy, it can have long-term adverse effects that negatively impact patient quality of life. Stereotactic radiosurgery and stereotactic body radiotherapy are advanced approaches to radiotherapy that can more precisely target radiation to tumors than traditional radiotherapy. The high degree of precision means that higher doses of radiation can be used compared with traditional radiotherapy and that healthy tissues surrounding a tumor are spared from damage caused by the radiation, which can reduce the long-term adverse effects of radiotherapy. Given the potential benefits of stereotactic radiosurgery and stereotactic body radiotherapy there are many clinical trials testing ways to incorporate these treatments into clinical cancer care. For example, stereotactic radiosurgery is increasingly being used after surgical removal of a brain metastasis (a tumor that has spread from another part of the body to the brain) because it was shown to lead to equally good overall survival with less neurocognitive deterioration compared with whole brain radiotherapy (197).
Another recent advance in radiotherapy is the emergence of hypofractionated radiotherapy, whereby patients receive fewer but higher doses of radiotherapy compared with the traditional course of radiotherapy. Thus, patients who have hypofractionated radiotherapy complete their radiotherapy over a shorter period of time and in fewer treatment sessions. Hypofractionated radiotherapy is increasingly being used in the treatment of early-stage breast cancer and localized prostate cancer because it was recently shown to be as effective as traditional courses of radiotherapy at reducing cancer recurrence after 10 years (198-199).
Typically, the only use of radiotherapy in the treatment of patients with metastatic cancer is to reduce or control symptoms of disease. However, several recent clinical trials have shown that radiotherapy targeted to the initial cancer from which tumors have metastasized can improve survival for patients who have metastatic tumors at a limited number of sites and are said to have oligometastatic disease. For example, two recent clinical trials have shown that adding prostate-targeted radiotherapy to standard treatment for metastatic prostate cancer significantly increased survival for patients who had limited metastatic disease (200-201). Similar benefits have been seen for patients with lung cancer who have oligometastatic disease (202-203).
Treatment with Cytotoxic Chemotherapy
Cytotoxic chemotherapy was the third type of treatment to become a pillar of cancer care (see Figure 10). The use of this mainstay of cancer treatment is constantly evolving as researchers develop new cytotoxic chemotherapeutics and identify new ways to use existing cytotoxic chemotherapeutics to improve survival and quality of life for patients.
A New Therapeutic for Patients with Acute Lymphoblastic Leukemia
In December 2018, the FDA approved a new therapeutic called calaspargase pegol-mknl (Asparlas) for use as part of a multiagent cytotoxic chemotherapy regimen for children, adolescents, and young adults ages 1 month to 21 years who have acute lymphoblastic leukemia (ALL).
More than 50 percent of those diagnosed with ALL each year in the United States are children, adolescents, and young adults under the age of 22. Most are treated with a combination of four, or even five, chemotherapeutics. One of the chemotherapeutics in the most commonly used combination works by depleting the patient’s body of a molecule called L-asparagine, which is one of the building blocks that cells use to create the proteins they need to function. ALL cells are unable to generate their own L-asparagine. Thus, depletion of this critical building block interferes with the ability of ALL cells to generate proteins and, ultimately, to survive.
Chemotherapeutics that work by depleting L-asparagine are asparagine-specific enzymes. In many cases, the enzyme is linked to a molecule called polyethylene glycol forming a pegaspargase. This slows down clearance of the enzyme from the patient’s body.
In calaspargase pegol-mknl, the linker used to attach the polyethylene glycol to the asparagine-specific enzyme is different from the linkers used in other pegaspargase chemotherapeutics. The new linker is even more stable than previous linkers, which further slows down clearance of the enzyme from a patient’s body (204). Thus, patients can go longer between doses of calaspargase pegol-mknl than they can between doses of other pegaspargase chemotherapeutics.
Treating Stomach Cancer in a New Way
In February 2019, the FDA approved a new use for Lonsurf, which is a single tablet that contains a combination of the cytotoxic chemotherapeutic trifluridine and a drug called tipiracil. The trifluridine – tipiracil combination tablet was approved for treating certain patients with stomach cancer, which is sometimes called gastric cancer, or gastroesophageal junction adenocarcinoma, which is cancer of the part of the esophagus that connects to the stomach. It was first approved by the FDA for treating advanced colorectal cancer in September 2015.
Trifluridine and tipiracil work together against cancer. Trifluridine causes damage to DNA in the rapidly multiplying cancer cells, which can ultimately trigger cell death; tipiracil prevents rapid breakdown of trifluridine, thereby maintaining adequate levels of the cytotoxic chemotherapeutic in the body.
Trifluridine damages DNA in a similar way to other cytotoxic chemotherapeutics called fluoropyrimidines, which have been used as a treatment for stomach cancer for decades. In a phase III clinical trial, the trifluridine – tipiracil combination tablet improved survival compared with placebo even for those patients who had stomach cancer or gastroesophageal junction adenocarcinoma that was no longer responding to treatment with fluoropyrimidine-containing chemotherapy regimens (205). On the basis of these results, the FDA approved Lonsurf for treating adult patients with metastatic stomach cancer or gastroesophageal junction adenocarcinoma that has progressed despite treatment with at least two other cytotoxic chemotherapy regimens, including one that includes a fluoropyrimidine.
Tailoring Cytotoxic Chemotherapy: Less Is Sometimes More
Treatment with cytotoxic chemotherapeutics can have adverse effects on patients. These effects can occur during treatment and continue long-term or they can appear months or even years later. As a result, health care providers and researchers are investigating whether less aggressive chemotherapy regimens can allow some patients the chance of an improved quality of life without an adverse effect on their survival.
In one recent phase III clinical trial involving frail or elderly patients with advanced stomach or esophageal cancer, researchers investigated how to optimize treatment with a combination of the cytotoxic chemotherapeutics oxaliplatin and capecitabine to be as effective as possible at slowing progression of the cancer while allowing the patients to maintain quality of life (206). They found that the lowest dose of oxaliplatin and capecitabine, which was 60 percent of the highest dose, was comparable to the highest dose in terms of delaying disease progression, but that it caused fewer adverse side effects and allowed patients to maintain a higher quality of life.
Another group for whom treatment de-escalation might be possible is patients with breast cancer who have an excellent response to chemotherapy given before surgery (neoadjuvant chemotherapy) (207). In a recent study, researchers found that patients who had no signs of invasive cancer in the breast tissue and lymph nodes removed during surgery following neoadjuvant chemotherapy (that is, patients who had a pathologic complete response) were less likely to have disease recurrence and were more likely to survive than those patients who did not have a pathologic complete response. The link between having a pathologic complete response after neoadjuvant chemotherapy and improved outcomes was seen regardless of whether the patient had additional chemotherapy after surgery. As this study was not a randomized clinical trial, further clinical trials are needed before this approach to treatment de-escalation can be considered for implementation in the clinic.
A third group for whom it might be appropriate to use less cytotoxic chemotherapy than is currently recommended is children with a rare type of liver cancer, called hepatoblastoma, who have had surgery to remove the tumor (208). About 100 children are newly diagnosed with hepatoblastoma each year in the United States. Among these children, about one-third have tumors that can be surgically removed as the initial treatment after diagnosis. Surgery has traditionally been followed by four cycles of a chemotherapy regimen comprising three cytotoxic chemotherapeutics – cisplatin, fluorouracil, and vincristine. Surgery followed by cytotoxic chemotherapy yields good outcomes for patients, with about 90 percent surviving 5 or more years after diagnosis. However, cytotoxic chemotherapy causes many adverse effects. In a recent small phase III clinical trial, 91 percent of patients with hepatoblastoma who had surgery at diagnosis and were then treated with two rather than four cycles of cisplatin, fluorouracil, and vincristine were alive 5 or more years after diagnosis (208). Thus, reducing postoperative chemotherapy might provide a way to reduce the risk of acute and long-term side effects from cytotoxic chemotherapy for certain patients with hepatoblastoma without an adverse effect on their survival. Results from another clinical trial suggest that it might be possible to postoperatively treat patients with hepatoblastoma with only cisplatin and then treat them with sodium thiosulfate to reduce their risk of hearing loss, which is one of the common adverse effects of cisplatin (209). Larger clinical trials are underway to confirm these data.
Treatment with Molecularly Targeted Therapy
Remarkable advances in our understanding of the biology of cancer, including the identification of numerous genetic mutations that fuel tumor growth in certain patients, set the stage for the new era of precision medicine, an era in which the standard of care for many patients is changing from a one-size-fits-all approach to one in which greater understanding of the individual patient and the characteristics of his or her cancer dictates the best treatment option for the patient (see
Understanding How Cancer Develops).
Therapeutics directed to the molecules influencing cancer cell multiplication and survival target the cells within a tumor more precisely than cytotoxic chemotherapeutics, which target all rapidly dividing cells, thereby limiting damage to healthy tissues. The greater precision of these molecularly targeted therapeutics tends to make them more effective and less toxic than cytotoxic chemotherapeutics. As a result, they are not only saving the lives of patients with cancer, but also allowing these individuals to have a higher quality of life.
In the 12 months spanning August 1, 2018 to July 31, 2019, the FDA approved 14 new molecularly targeted anticancer therapeutics (see Table 5). During this period, they also approved four previously approved molecularly targeted anticancer therapeutics for treating additional types of cancer.
Targeting Cancers Based on Tumor Biomarker, Not Tumor Origin
One of the most significant precision medicine advances in the 12 months spanning this report was the first FDA approval of a molecularly targeted therapeutic to treat cancer based on the presence of a specific genetic biomarker in the tumor irrespective of the site at which the tumor originated. The therapeutic, larotrectinib (Vitrakvi), was approved by the FDA in November 2018 for treating children and adults who have solid tumors that test positive for the NTRK gene fusion biomarker.
The approval of larotrectinib for use in a tissue-agnostic way followed several decades of basic, translational, and clinical research (see Figure 12).
Larotrectinib targets three related proteins called TRKA, TRKB, and TRKC. The genes NTRK1, NTRK2, and NTRK3 provide the code that cells use to make these proteins.

Structural changes in chromosomes that involve the three NTRK genes and lead to the production of NTRK gene fusions, and subsequently to TRK fusion proteins, have been identified in a diverse array of cancer types that occur in adults and children (166) (see sidebar on Genetic Mutations). These include many rare types of cancer, including mammary analogue secretory carcinoma of the salivary gland, infantile fibrosarcoma, and cholangiocarcinoma. Overall, researchers estimate that NTRK gene fusions fuel the growth of up to 1 percent of all solid tumors.
Larotrectinib was approved after it was shown in three basket trials (see Figure 9) that 75 percent of patients treated with the molecularly targeted therapeutic had complete or partial tumor shrinkage (166). There were 17 types of cancer represented among the group of 55 patients enrolled in the three trials, with the most common being mammary analogue secretory carcinoma of the salivary gland. Tumor shrinkage was seen across cancer types. Thus, the approval provides a new treatment option and new hope to patients with a wide range of types of cancer, including children with soft tissue sarcoma, such as Emma Levine, and adults with salivary gland tumors, such as Keith Taggart.

Despite major advances in the treatment of breast cancer, this disease is the second-leading cause of cancer-related death for women in the United States (2). Recent FDA decisions have the potential to power more progress against breast cancer because they have provided new molecularly targeted therapeutic treatment options for certain patients with the disease.
Providing New Options for Patients with Breast Cancer
Despite major advances in the treatment of breast cancer, this disease is the second-leading cause of cancer-related death for women in the United States (2). Recent FDA decisions have the potential to power more progress against breast cancer because they have provided new molecularly targeted therapeutic treatment options for certain patients with the disease.
For patients with breast cancer, one factor determining what treatment options should be considered is the presence or absence of three tumor biomarkers, two hormone receptors and HER2. About 70 percent of breast cancers diagnosed in the United States are characterized as hormone receptor – positive and HER2-negative (210). Potential treatment options for these patients include therapeutics such as tamoxifen, which works by preventing the hormone estrogen from attaching to its receptor; letrozole, which works by lowering the level of estrogen in the body; and fulvestrant, which works by reducing the number of receptors for estrogen to bind to and by preventing estrogen from attaching to its receptor. Treatment with these therapeutics is often called endocrine therapy.
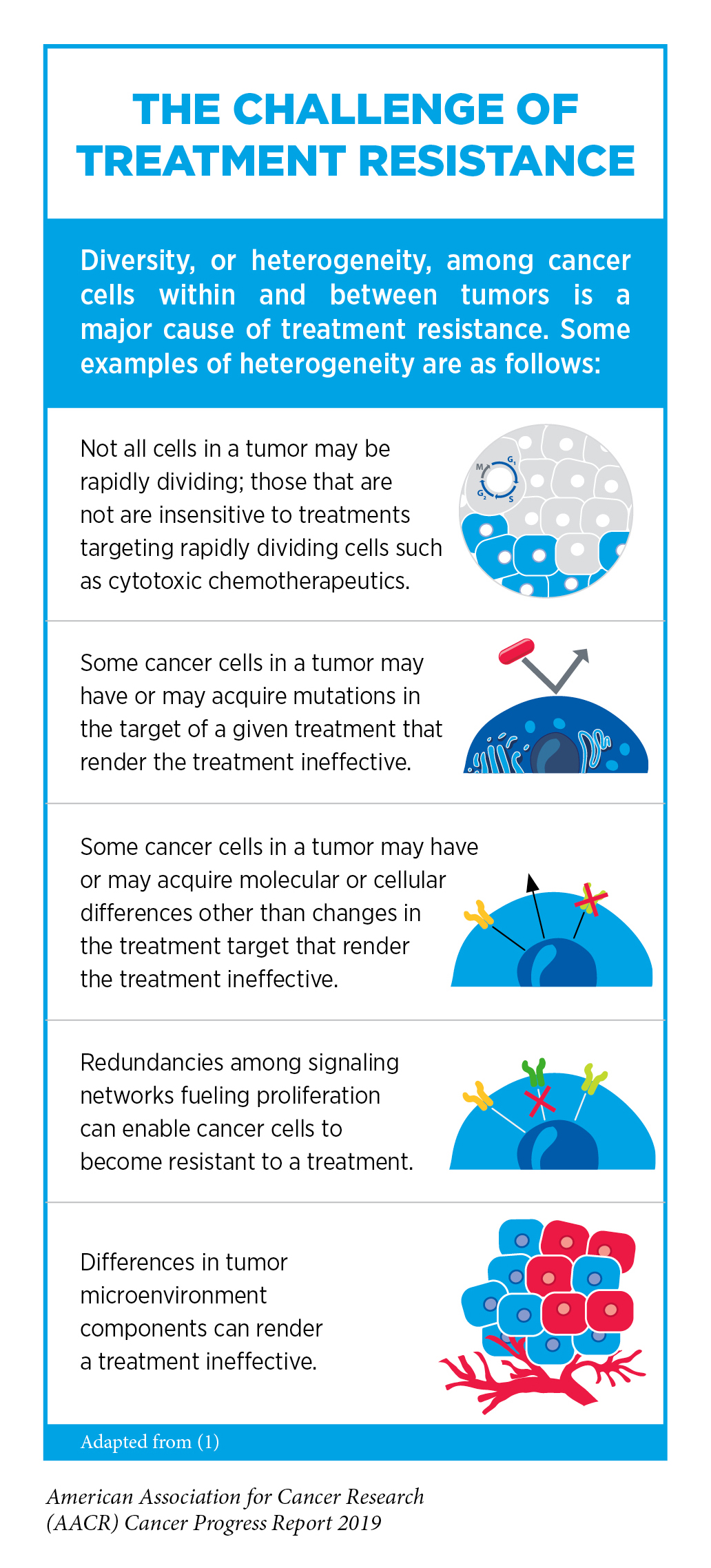
Unfortunately, most advanced, hormone receptor – positive breast cancers that initially respond to endocrine therapy eventually progress because they have become treatment resistant (see sidebar on The Challenge of Treatment Resistance). In May 2019, the FDA approved the molecularly targeted therapeutic alpelisib (Piqray) as a new treatment option to help address this challenge.
Alpelisib works by blocking the function of phosphatidylinositol 3-kinase (PI3K) alpha, which has an important role in driving cell multiplication and survival. Research has shown that mutations in the PIK3CA gene, which provides the code that cells use to make the PI3K-alpha protein, promote the multiplication and survival of about 40 percent of hormone receptor – positive, HER2-negative breast cancers (211).
Alpelisib was approved by the FDA for use in combination with fulvestrant for treating men and postmenopausal women who have advanced or metastatic, hormone receptor – positive, HER2-negative breast cancer that tests positive for PIK3CA mutations and has progressed during or after endocrine therapy. This approval was based on results from a phase III clinical trial that showed that adding alpelisib to fulvestrant almost doubled the time before disease progression (211).

At the same time that the FDA made the decision about alpelisib, it approved the therascreen PIK3CA RGQ PCR Kit as a companion diagnostic to test patients for PIK3CA mutations (see sidebar on Companion Diagnostics). This companion diagnostic can be used to test either tumor tissue or circulating tumor DNA isolated from blood samples, which can reduce the invasiveness of testing (see Liquid Biopsies).
Another FDA decision that provided an advance against breast cancer was the October 2018 approval of the molecularly targeted therapeutic talazoparib (Talzenna) for treating patients with metastatic or locally advanced, HER2-negative breast cancer who have inherited a known or suspected cancer-associated BRCA1 or BRCA2 mutation. At the same time, the FDA approved using the BRACAnalysis CDx test as a companion diagnostic to identify patients who are eligible for talazoparib.
About 5 percent of all breast cancers diagnosed in the United States are attributable to an inherited BRCA1 or BRCA2 mutation (210).
The reason that the presence of a BRCA1 or BRCA2 mutation is relevant relates to the way that talazoparib works. Talazoparib targets poly ADP-ribose polymerase (PARP) proteins. Decades of basic research have shown that a key function of both PARP and BRCA proteins is repairing damaged DNA. Although they work in different DNA repair pathways, the pathways are interrelated and disruption to both pathways can ultimately trigger cell death. As a result, cancer cells harboring cancer-associated BRCA gene mutations that disable the ability of BRCA proteins to repair damaged DNA are particularly susceptible to PARP inhibitors, which work, at least in part, by blocking the DNA repair function of PARP proteins.
The approval of talazoparib was based on results from a phase III clinical trial that showed that treatment with the molecularly targeted therapeutic significantly increased the time to disease progression compared with treatment with a cytotoxic chemotherapeutic (212).
Given the benefits of talazoparib and another PARP inhibitor called olaparib (Lynparza) for patients with metastatic or locally advanced, HER2-negative breast cancer who have inherited a known or suspected cancer-associated BRCA1 or BRCA2 mutation, patients should talk with their health care providers about whether they are at high risk for having inherited one of these mutations and whether genetic testing is right for them. This is important because a recent study found that half of patients with breast cancer who were at high risk for having inherited a known or suspected cancer-associated BRCA1 or BRCA2 mutation had not had a genetic test (213).
Treating Bladder Cancer with a Novel Therapeutic
Bladder cancer is the sixth most commonly diagnosed cancer in the United States, with more than 80,000 new cases expected to be diagnosed in 2019 (2).
More than 90 percent of bladder cancers diagnosed in the United States are classified as urothelial carcinomas because they arise in cells that comprise the transitional cell urothelium that lines the bladder. Research has shown that up to 30 percent of urothelial carcinomas have a mutation in one of the four FGFR genes, with the most common being mutations in the FGFR3 gene (214). These mutations help promote the multiplication and survival of the cells within the urothelial carcinoma.

Erdafitinib (Balversa) targets FGFR proteins. In April 2019, it was approved for treating patients who have locally advanced or metastatic urothelial carcinoma that tests positive for FGFR2 or FGFR3 genetic mutations and that has progressed during or after treatment with a platinum-based cytotoxic chemotherapeutic. The approval was granted after it was shown in a phase II clinical trial that more than one-third of the patients who received erdafitinib had complete or partial tumor shrinkage.
At the same time that the FDA made the decision about erdafitinib, it approved the therascreen FGFR RGQ RT-PCR Kit as a companion diagnostic to identify patients with urothelial carcinoma who are eligible for the molecularly targeted therapeutic, such as Gary Price.
Adding Precision to the Treatment of Blood Cancers

Cancers that arise in blood-forming tissues, such as the bone marrow, or in cells of the immune system are called blood cancers, or hematologic cancers. In the 12 months covered by this report, the FDA has made numerous decisions that are transforming the lives of patients with a wide array of hematologic cancers, including approving seven new molecularly targeted therapeutics for patients with some of these diseases (see sidebar on Recent Advances against Blood Cancers).
Acute myeloid leukemia (AML) is the most commonly diagnosed type of leukemia in the United States, with 21,450 new cases anticipated in 2019 (2). It is also the type of leukemia with the lowest overall 5-year relative survival rate, 28 percent (8). In November 2018, the FDA made three decisions that provided new treatment options for defined groups of patients with this devastating disease.
One of these decisions was the approval of gilteritinib (Xospata), which targets FLT3. Mutations in the FLT3 gene promote the multiplication and survival of AML cells in 25 percent to 30 percent of cases, and patients with this type of AML have particularly poor outcomes (215). Gilteritinib was approved for treating adults who have AML that tests positive for a FLT3 mutation and that has not responded to or has relapsed after initial treatment. The approval was based on early results from a phase III clinical trial that showed that 21 percent of patients who received gilteritinib had complete remission – this means they had no evidence of disease and full recovery of blood counts – or complete remission with partial hematologic recovery – meaning no evidence of disease and partial recovery of blood counts. Subsequent results from the trial showed that gilteritinib also almost doubled median overall survival compared with standard chemotherapy regimens (216).
At the same time that the FDA made the decision about gilteritinib, it approved expanding the use of the LeukoStrat CDx FLT3 Mutation Assay to allow it to be used as a companion diagnostic to identify patients with FLT3 mutation – positive AML who are eligible for treatment with the new molecularly targeted therapeutic.
The FDA approved a second new molecularly targeted therapeutic for the treatment of AML in November 2018, glasdegib (Daurismo). Glasdegib targets a protein called Smoothened, which is part of a signaling pathway implicated in driving AML progression (217). Glasdegib was approved for use in combination with a low dose of the cytotoxic chemotherapeutic cytarabine for treating patients newly diagnosed with AML who are 75 or older or who have other chronic health conditions or diseases (comorbidities) that prevent them from having standard intensive cytotoxic chemotherapy. The approval was based on results from a phase II clinical trial that showed that adding glasdegib to low-dose cytarabine almost doubled median overall survival (218).
In November 2018, the FDA also approved expanding the use of the molecularly targeted therapeutic venetoclax (Venclexta) to include the treatment of patients newly diagnosed with AML who are 75 or older or who have comorbidities that prevent them from having standard intensive cytotoxic chemotherapy. Venetoclax targets the protein BCL-2, which promotes cell survival by preventing cells from undergoing a natural self-destruct process called apoptosis. Research has shown that levels of BCL-2 are frequently elevated in several types of leukemia cells, including AML cells and chronic lymphocytic leukemia (CLL) cells, and that it promotes the survival of these cells (219). By blocking BCL-2, venetoclax triggers the leukemia cells to die by apoptosis.
The approval of venetoclax for treating AML is for use of the molecularly targeted therapeutic in combination with any one of the cytotoxic chemotherapeutics azacitidine, decitabine, or low-dose cytarabine. It was based on results from two phase I/II clinical trials. In one of the trials, 67 percent of patients who received venetoclax and azacitidine had complete remission and 54 percent of patients who received venetoclax and decitabine had complete remission (219). In the other trial, 21 percent of patients who received venetoclax and low-dose cytarabine had complete remission. Venetoclax was first approved by the FDA for treating certain patients with CLL in April 2016.
CLL arises in immune cells called B cells, as do several other types of blood cancer, including small lymphocytic lymphoma (SLL). CLL and SLL are essentially the same disease, but have different names depending on where in the body the cancer cells accumulate. CLL cells are found mostly in the blood and bone marrow, whereas SLL cells are found mostly in the lymph nodes. In September 2018, the FDA approved a new molecularly targeted therapeutic for treating patients with CLL/SLL, duvelisib (Copiktra). Duvelisib targets PI3K-delta and PI3K-gamma, which are protein components of signaling pathways that have a key role in promoting the survival and expansion of CLL/SLL cells (220). Duvelisib was approved by the FDA for treating patients with CLL/SLL whose disease has progressed after they have received at least two other types of treatment. The approval was based on results from a phase III clinical trial that showed that a significantly greater proportion of patients responded to treatment with duvelisib compared with the standard treatment, ofatumumab (Arzerra) (220). The median time to disease progression was also longer among those who received duvelisib.
At the same time as approving duvelisib for CLL/SLL, the FDA approved the therapeutic for treating certain patients with follicular lymphoma, a type of non-Hodgkin lymphoma that arises in B cells. The approval of duvelisib for treating patients with follicular lymphoma whose disease has progressed after they have received at least two other types of treatment was based on results from a phase III clinical trial. The results showed that 42 percent of patients whose disease was not responding to standard treatments had partial or complete tumor shrinkage following treatment with duvelisib.
Multiple myeloma is one of the most commonly diagnosed blood cancers in the United States, with 32,110 new cases expected to be diagnosed in 2019 (2). In recent years, the development and FDA approval of new therapeutics – including proteasome inhibitors like bortezomib (Velcade) and carfilzomib (Kyprolis), immunomodulatory agents like lenalidomide (Revlimid), and immunotherapeutics like the CD38-targeted daratumumab (Darzalex) – have improved outcomes for patients. Despite the advances, many patients whose disease initially responds to the new therapeutics eventually relapse due to treatment resistance.
In July 2019, the FDA approved a new molecularly targeted therapeutic called selinexor (Xpovio) for treating patients with multiple myeloma whose disease has relapsed subsequent to, or never responded to, treatment with at least two proteasome inhibitors, at least two immunomodulatory agents, and a CD38-targeted immunotherapeutic. Selinexor targets a protein called XPO1, which is found at elevated levels in multiple myeloma cells. XPO1 helps move proteins out of a part of the cell called the nucleus. It is particularly linked to moving proteins that suppress tumor growth out of the nucleus. When selinexor targets XPO1, it forces these proteins to be retained in the nucleus where they can act to suppress tumor growth. The approval of selinexor was based on results from a phase II clinical trial that showed that 25 percent of heavily pretreated patients responded to treatment with the new molecularly targeted therapeutic.
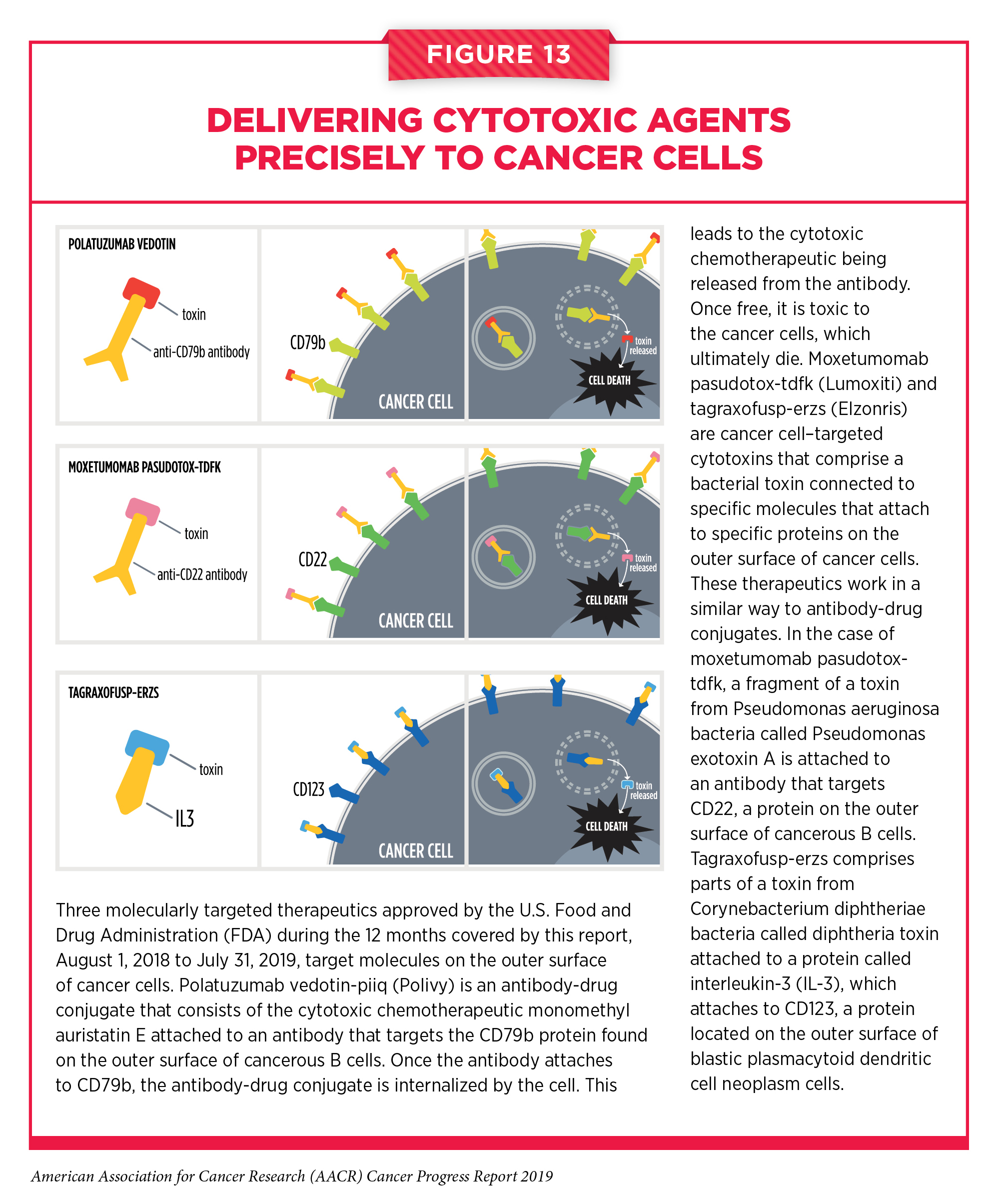
All of the molecularly targeted therapeutics for treating blood cancers that have been discussed above target specific molecules inside cancer cells. Three other molecularly targeted therapeutics approved by the FDA for treating blood cancers during the 12 months covered by this report target molecules on the outer surface of cancer cells (see Figure 13). Polatuzumab vedotin-piiq (Polivy) is the most recently approved of these three therapeutics. It is an antibody-drug conjugate. Antibody-drug conjugates use an antibody to deliver an attached cytotoxic chemotherapeutic directly to the cancer cells with the antibody’s target on their surfaces. Once the antibody attaches to its target on the surface of a cancer cell, the antibody-drug conjugate is internalized by the cell. This leads to the cytotoxic chemotherapeutic being released from the antibody. Once free, it is toxic to the cancer cells, which ultimately die. The precision of antibody targeting reduces the side effects of the cytotoxic chemotherapeutic compared with traditional systemic delivery.
In the case of polatuzumab vedotin-piiq, the cytotoxic agent monomethyl auristatin E is attached to a CD79b-targeted antibody. CD79b is found on the surface of immune cells called B cells, both normal B cells and those that become cancerous. Diffuse large B-cell lymphoma is an aggressive type of non-Hodgkin lymphoma that arises in B cells. It is the most common type of non-Hodgkin lymphoma diagnosed in the United States. Polatuzumab vedotin-piiq was approved for use in combination with the cytotoxic chemotherapeutic bendamustine and the immunotherapeutic rituximab for treating adults who have large B-cell lymphoma that has not responded to or has relapsed after two other treatments. The June 2019 approval was based on results from a phase Ib/II clinical trial that showed that 40 percent of patients who received all three of the anticancer therapeutics had complete tumor shrinkage compared with 18 percent of those who received only bendamustine and rituximab.
The other two therapeutics that target molecules on the outer surface of cancer cells, moxetumomab pasudotox-tdfk (Lumoxiti) and tagraxofusp-erzs (Elzonris), are cancer cell – targeted cytotoxins. These therapeutics work in a similar way to antibody-drug conjugates. They comprise two parts with different functions. As in antibody-drug conjugates, one part attaches to a specific molecular target on the outer surface of certain cancer cells, but the part in these therapeutics that kills cancer cells is a bacterial toxin, not a cytotoxic chemotherapeutic.
In the case of moxetumomab pasudotox-tdfk, the cytotoxin is a fragment of a toxin from Pseudomonas aeruginosa bacteria called Pseudomonas exotoxin A and the targeting portion attaches to CD22, a molecule on the outer surface of immune cells called B cells. Thus, CD22 is on the surface of most blood cancers that arise in B cells, including a rare type of slow-growing leukemia called hairy cell leukemia. The first treatment for most patients with hairy cell leukemia is the cytotoxic chemotherapeutic cladribine. Although this treatment often leads to durable remission, 30 percent to 40 percent of patients relapse after 5 to 10 years (221).
In September 2018, moxetumomab pasudotox-tdfk was approved for treating patients who have hairy cell leukemia that has not responded to or has relapsed after two treatments, including cladribine or another cytotoxic chemotherapeutic that works in the same way. The approval was based on results from a phase III clinical trial that showed that 30 percent of the patients who received moxetumomab pasudotox-tdfk had a durable complete response, which was defined as maintenance of hematologic remission for more than 180 days (222). Even though there are only about 1,000 new cases of hairy cell leukemia diagnosed each year in the United States (223), this approval provides a new treatment option and new hope for patients with the disease, such as Randy Surratt.

Tagraxofusp-erzs is a CD123-targeted cytotoxin. It comprises parts of a toxin from Corynebacterium diphtheriae bacteria called diphtheria toxin and a protein called interleukin-3 (IL-3), which attaches to CD123. CD123 is a molecule on the surface of blastic plasmacytoid dendritic cell neoplasm cells. Blastic plasmacytoid dendritic cell neoplasm is a rare type of blood cancer that is highly aggressive. Although many patients respond to treatment with a combination of cytotoxic chemotherapeutics, this treatment strategy ultimately fails to control the disease. The median survival of patients diagnosed with blastic plasmacytoid dendritic cell neoplasm is 8 to 14 months (224).
In December 2018, tagraxofusp-erzs became the first treatment approved by the FDA specifically for blastic plasmacytoid dendritic cell neoplasm. It was approved for treating patients who are age 2 or older after it was shown in an early-stage clinical trial that seven of 13 patients (54 percent) with previously untreated blastic plasmacytoid dendritic cell neoplasm who received the new CD123-targeted cytotoxin had a complete response (defined as disappearance of disease at each site of initial disease) or a clinical complete response (defined as a complete response with residual skin abnormality not indicative of active disease). A subsequent report on the trial that included results from a larger number of patients showed that 21 of 29 patients (72 percent) with previously untreated blastic plasmacytoid dendritic cell neoplasm had a complete response or a clinical complete response (224). In addition, 52 percent of the patients were alive 24 months after starting treatment with tagraxofusp-erzs.
Increasing Options for Patients with Prostate Cancer
Prostate cancer is the most commonly diagnosed cancer among men in the United States (2). It is also the second-leading cause of cancer death for U.S. men.
Most men who die from prostate cancer have metastatic disease. Therefore, one goal of prostate cancer researchers is to identify new ways to increase the time before early-stage disease progresses and becomes metastatic. The molecularly targeted therapeutic darolutamide (Nubeqa) recently became the third treatment approved by the FDA based on its ability to do the above.
At the time of diagnosis, the growth of most prostate cancers is fueled by hormones called androgens. Androgens, such as testosterone, attach in a lock-and-key fashion to androgen receptors on individual prostate cancer cells, stimulating the cancer cells to multiply and survive. This knowledge led researchers to develop treatments that lower androgen levels in the body or stop androgens from attaching to androgen receptors. This approach to prostate cancer treatment is called androgen-deprivation therapy. It is an important part of care for many men with the disease. Unfortunately, most prostate cancers that initially respond to androgen-deprivation therapy eventually begin to grow again. At this point they are said to be castration resistant.
Even though the approaches to androgen-deprivation therapy that have become the mainstay of prostate cancer treatment (bilateral orchiectomy or treatment with a gonadotropin-releasing hormone analogue agonist or antagonist) reduce androgen levels in the body, they do not eliminate these hormones completely. As a result, castration-resistant prostate cancer growth is still often fueled by androgens. Therefore, researchers have begun to develop a new generation of therapeutics that more effectively deprive prostate cancer of androgens. The first of these therapeutics, abiraterone (Zytiga) and enzalutamide (Xtandi), were approved by the FDA for treating men with metastatic castration-resistant prostate cancer in 2011 and 2012, respectively. Then, in 2018, the FDA approved enzalutamide and apalutamide (Erleada) for treating men with nonmetastatic castration-resistant prostate cancer.
The July 2019 approval of darolutamide provides men with nonmetastatic castration-resistant prostate cancer a third treatment option. The approval of this therapeutic was based on results from a phase III clinical trial that showed that adding darolutamide to standard androgen-deprivation therapy increased the time before prostate cancer metastasized by almost 2 years (225).
Expanding Treatment Options for Lung Cancer Patients
Lung cancer is the second most commonly diagnosed cancer in the United States, with more than 228,150 new cases expected to be diagnosed in 2019 (2). About 85 percent of lung cancers diagnosed in the United States are classified as non-small cell lung cancers (NSCLC).
In recent years, researchers have significantly increased our understanding of the genetic changes that fuel NSCLC growth in certain patients and have developed therapeutics that target some of these changes (226).
One of the genes most frequently mutated in NSCLC cancer is EGFR (226). Dacomitinib (Vizimpro) is a new EGFR-targeted therapeutic approved by the FDA in September 2018. It was approved as an initial treatment for patients with metastatic NSCLC that tests positive for certain EGFR mutations, either an EGFR exon 19 deletion or the exon 21 L858R mutation. The approval was based on results from a phase III clinical trial that showed that treatment with dacomitinib significantly increased the time to disease progression compared with gefitinib (Iressa), which is the EGFR-targeted therapeutic most commonly used to initially treat patients with metastatic NSCLC that tests positive for EGFR mutations (227). Such second-line treatments are important because many NSCLCs that initially respond to molecularly targeted therapeutics eventually progress due to the development of treatment resistance.
In November 2018, the FDA approved a molecularly targeted therapeutic called lorlatinib (Lorbrena), providing a new option to help patients with NSCLC fueled by mutations in the ALK gene to address the challenge of treatment resistance.
The ALK gene is another gene frequently altered in NSCLC. Crizotinib (Xalkori) was the first ALK-targeted therapeutic to be approved by the FDA, in August 2011. It was followed by ceritinib (Zykadia), which was approved in April 2014; alectinib (Alecensa), which was approved in December 2015; and brigatinib (Alunbrig), which was approved in April 2017.
In many cases, resistance to crizotinib, ceritinib, alectinib, and brigatinib emerges because NSCLC cells acquire additional ALK mutations. Research has shown that the second-generation ALK-targeted therapeutics – ceritinib, alectinib, and brigatinib – can inhibit most of the ALK mutations that drive resistance to crizotinib and the third-generation ALK-targeted therapeutic “lorlatinib” can inhibit most of the ALK mutations that drive resistance to the second-generation ALK-targeted therapeutics (228).
Lorlatinib was approved for treating patients who have metastatic NSCLC driven by mutations in the ALK gene and whose disease has progressed despite treatment with crizotinib and at least one other ALK-targeted therapeutic for metastatic disease; treatment with alectinib as the first ALK-targeted therapeutic for metastatic disease; or treatment with ceritinib as the first ALK-targeted therapeutic for metastatic disease. The approval was based on results from a phase II clinical trial that showed that 48 percent of patients with ALK mutation – positive metastatic NSCLC who had previously been treated with one or more ALK-targeted therapeutics had complete or partial tumor shrinkage following treatment with lorlatinib (229).
As researchers seek to drive more progress against ALK-driven NSCLC, one area of intensive research investigation is determining the optimal sequence in which to use the five FDA-approved ALK-targeted therapeutics to provide the maximum benefit for patients.
Blocking the Blood Supply to Liver Cancer
Research has shown that many solid tumors need to establish their own blood supply and lymphatic vessel network to grow and survive. Identification of molecules that control the growth of new blood and lymphatic vessels within a tumor led to the development of therapeutics that specifically block them. These therapeutics are referred to as antiangiogenic agents.
From 2004 to 2015, the FDA approved 11 new antiangiogenic agents. These therapeutics are now approved for treating a wide array of cancer types (see
Supplemental Table 2a, 2b, 2c, 2d). In the 12 months covered by this report, the FDA expanded the use of three of the antiangiogenic agents to include the treatment of a new type of cancer, cabozantinib (Cabometyx), lenvatinib (Lenvima), and ramucirumab (Cyramza).
All three of the approvals have provided new treatment options for certain patients with the most common type of liver cancer, hepatocellular carcinoma. New approaches to treatment are urgently needed because the 5-year relative survival rate for liver cancer is 18 percent, which is one of the lowest 5-year relative survival rates for any type of cancer (2).
Most patients diagnosed with hepatocellular carcinoma that cannot be removed by surgery are initially treated with the antiangiogenic agent sorafenib (Nexavar). In August 2018, the FDA approved lenvatinib as an alternative treatment option for these patients. The approval was based on results from a phase III clinical trial that showed that overall survival among patients with hepatocellular carcinoma who were treated with lenvatinib was no worse than overall survival among patients who were treated with sorafenib (230).
Unfortunately, not all patients with hepatocellular carcinoma benefit from treatment with sorafenib or lenvatinib. Moreover, most patients whose tumors initially respond to these antiangiogenic agents eventually have disease progression.
In the first half of 2019, cabozantinib and ramucirumab were approved by the FDA for treating patients who have hepatocellular carcinoma that has progressed despite treatment with sorafenib. The approvals were based on results from phase III clinical trials that showed that the antiangiogenic agents improved overall survival compared with placebo (231).
Treatment with Immunotherapy

Cancer immunotherapeutics work by unleashing the power of a patient’s immune system to fight cancer the way it fights pathogens like the virus that causes influenza and the bacterium that causes strep throat. Not all immunotherapeutics work in the same way (see sidebar on
How Immunotherapeutics Work).
The use of immunotherapeutics in the treatment of cancer is referred to as cancer immunotherapy. In recent years, it has emerged as the fifth pillar of cancer care and is one of the most exciting new approaches to cancer treatment that has entered the clinic. This is in part because some patients with metastatic disease who have been treated with these revolutionary anticancer treatments have had remarkable and durable responses. For example, recent long-term results from a clinical trial testing the immunotherapeutic pembrolizumab (Keytruda) as an initial treatment for patients with advanced NSCLC showed that 23 percent lived 5 or more years, which stands in stark contrast to the historical 5-year relative survival rate for patients with advanced NSCLC of about 5 percent (232).
Unfortunately, at present, only a minority of patients have such dramatic responses. In addition, current FDA-approved immunotherapeutics are not effective against all types of cancer. Identifying ways to increase the number of patients for whom treatment with an immunotherapeutic yields a remarkable and durable response is an area of intensive basic and clinical research.

Fortunately, our scientific understanding of the immune system and how it interacts with cancer cells is rapidly increasing, and there are already clinical trials underway testing many novel immunotherapeutics and testing new ways to use those immunotherapeutics that we already have, such as combinations of those previously approved (233). One of the most promising advances is the development of a type of immunotherapy called adoptive T-cell therapy, which boosts the ability of the immune system to eliminate cancer cells (234) (see sidebar on Types of Adoptive T-cell Therapy). As of July 31, 2019, two of these new types of immunotherapy had been approved by the FDA, axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah). Both are a type of chimeric antigen receptor (CAR) T-cell therapy and are approved for treating certain patients with ALL and non-Hodgkin lymphoma. Early results from several small clinical trials suggest that researchers may have overcome some of the challenges to successfully developing CAR T-cell therapy for patients with certain solid tumors (235). However, more data from larger clinical trials will be needed to determine whether these new treatments provide significant benefit to patients and whether they have any long-term or late effects.
Clearly, the new immunotherapeutics and treatment strategies that are on the horizon hold extraordinary promise for the future. Here, we focus on new immunotherapeutics that were approved by the FDA in the 12 months covered by this report, August 1, 2018 to July 31, 2019, and previously approved immunotherapeutics that were approved for use against additional types of cancer during the same period.
Releasing the Brakes on the Immune System
Research has shown that immune cells called T cells are naturally capable of destroying cancer cells. It has also shown that some tumors evade destruction by T cells because they have high levels of proteins that attach to and trigger “brakes” on T cells, stopping the T cells from attacking the tumor. These brakes, which are proteins on the surface of T cells, are called immune checkpoint proteins.

This knowledge led to the development of immunotherapeutics that release certain T-cell brakes (see
Figure 14). These immunotherapeutics are called checkpoint inhibitors. Two of the researchers whose work was pivotal to the identification of immune checkpoint proteins and their function, and to the development of checkpoint inhibitors, James P. Allison, PhD, and Tasuku Honjo, MD, PhD, were recognized with the 2018 Nobel Prize in Physiology or Medicine for their discoveries of cancer therapy by inhibition of negative immune regulation.
The first checkpoint inhibitor to be approved by the FDA was ipilimumab (Yervoy), in March 2011. This immunotherapeutic targets the immune checkpoint protein CTLA-4, protecting it from the proteins that attach to it and trigger it to put the brakes on cancer-cell killing by T cells.
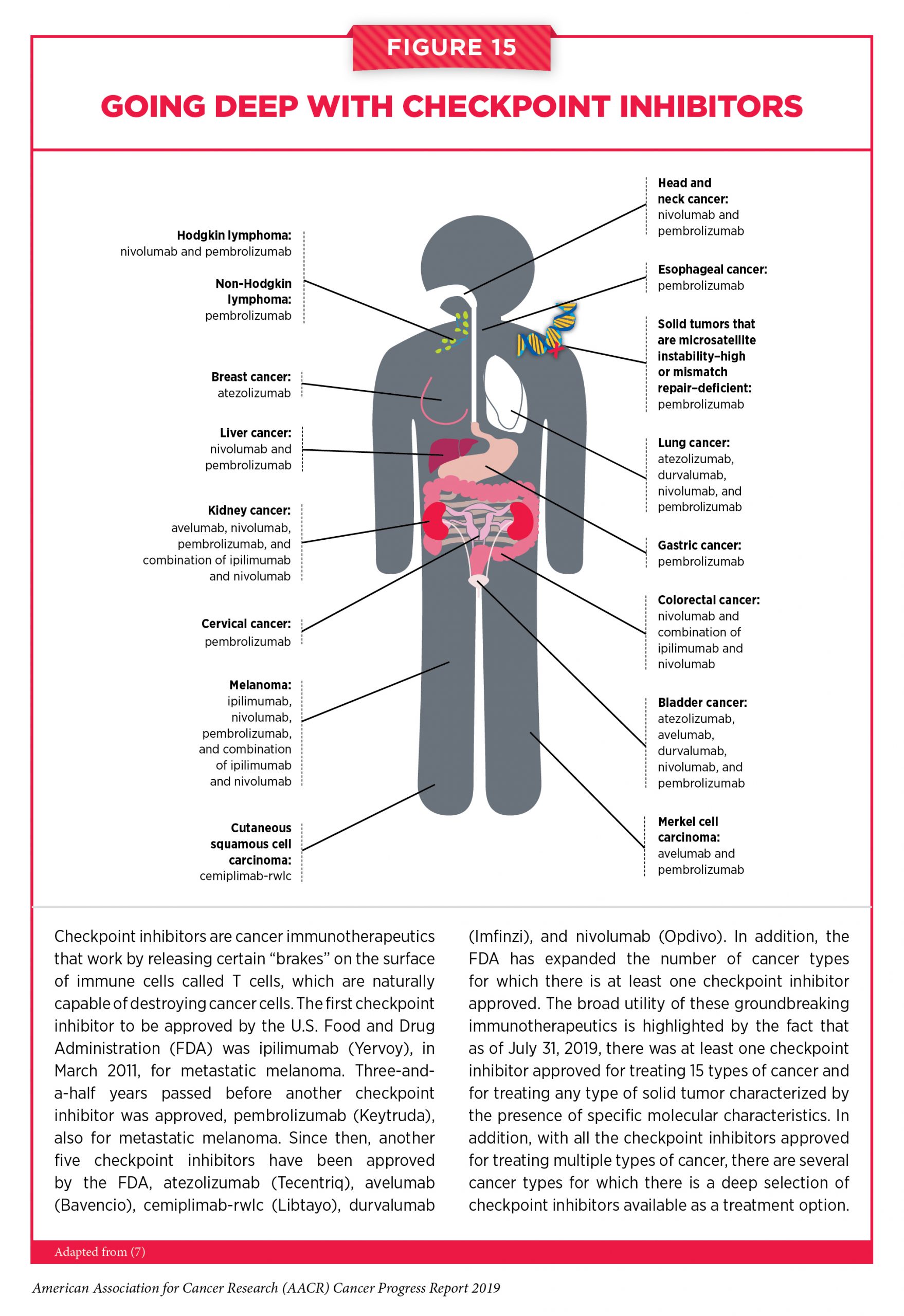
Since the first approval of ipilimumab for treating patients with metastatic melanoma, the use of checkpoint inhibitors in the treatment of cancer has rapidly expanded. The FDA has approved six other checkpoint inhibitors, all of which release a different T-cell braking system compared with ipilimumab. They target either the immune checkpoint protein PD-1 or PD-L1, which is one of the proteins that applies the PD-1 brake on T cells. The FDA has also approved these groundbreaking immunotherapeutics for an increasingly broad array of cancer types. During the 12 months spanning this report, August 1, 2018 to July 31, 2019, the FDA approved one checkpoint inhibitor for the first time – cemiplimab-rwlc (Libtayo) – and expanded the uses of four of the previously approved checkpoint inhibitors – avelumab (Bavencio), durvalumab (Imfinzi), nivolumab (Opdivo), and pembrolizumab (Keytruda) – to include the treatment of additional types of cancer. As of July 31, 2019, there was at least one checkpoint inhibitor approved for treating 15 types of cancer and for treating any type of solid tumor characterized by the presence of specific molecular characteristics (see Figure 15).

Cemiplimab-rwlc became the seventh checkpoint inhibitor approved by the FDA in September 2018. It was approved for treating patients like Harold Sokoloff who have metastatic cutaneous squamous cell carcinoma or locally advanced cutaneous squamous cell carcinoma that cannot be treated with curative surgery or curative radiation.
Cutaneous squamous cell carcinoma is the second most common type of skin cancer diagnosed in the United States. About 700,000 people were treated for the disease in 2012, which is the most recent year for which there are data (245). Most patients are cured by surgery and/or radiation. The emergence of advanced disease is rare. However, if it does occur, it can be difficult to treat and there were no therapeutics approved specifically to treat patients who were diagnosed with advanced cutaneous squamous cell carcinoma until the approval of cemiplimab-rwlc. The approval was based on results from two small clinical trials. Overall, 47 percent of the patients who received cemiplimab-rwlc had complete or partial tumor shrinkage. Most of these patients were continuing to gain benefit from the new checkpoint inhibitor at the time of its approval (246).
March 2019 marked another milestone in the era of checkpoint inhibitors. The FDA approved expanding the use of atezolizumab to include treating adults who have metastatic triple-negative breast cancer that tests positive for PD-L1 protein and adults who have locally advanced triple-negative breast cancer that cannot be removed by surgery and tests positive for PD-L1 protein. The approval was for the use of the checkpoint inhibitor in combination with the cytotoxic chemotherapeutic nab-paclitaxel (Abraxane). At the same time, the FDA approved the Ventana PD-L1 Assay as a companion diagnostic to identify patients with PD-L1 – positive, triple-negative breast cancer.
Triple-negative breast cancer accounts for about 12 percent of breast cancer cases diagnosed in the United States each year (210). Breast cancers are classified as triple-negative if they test negative for hormone receptors and the protein HER2. Until the approval of atezolizumab, cytotoxic chemotherapeutics were the only systemic treatment options for patients with triple-negative breast cancer.

This groundbreaking approval was based on results from a phase III clinical trial that showed that adding atezolizumab to nab-paclitaxel significantly increased the time before disease progression compared with placebo and nab-paclitaxel (247). Early results from the trial suggest that treatment with atezolizumab and nab-paclitaxel also improves overall survival compared with placebo and nab-paclitaxel. Although more time is needed to determine the extent of the survival benefit, atezolizumab has already transformed the lives of many patients with triple-negative breast cancer, such as Eva Joseph.
Atezolizumab was approved for treating an additional type of cancer in March 2019. It was approved for use in combination with two cytotoxic chemotherapeutics, carboplatin and etoposide, for the initial treatment of adults diagnosed with extensive-stage small-cell lung cancer (SCLC). SCLC accounts for about 15 percent of lung cancers diagnosed each year in the United States (2). Most patients are diagnosed with extensive-stage disease, which means the cancer has spread beyond the lung, or the area between the lungs or the lymph nodes above the collarbone to other parts of the body. Even with treatment, which is commonly a combination of the cytotoxic chemotherapeutics carboplatin or cisplatin and etoposide, median survival is about 10 months. The approval of atezolizumab was based on results from a phase III clinical trial that showed that adding atezolizumab to standard treatment with carboplatin and etoposide significantly improved median overall survival (248).
In addition to the approval of atezolizumab for the initial treatment of adults diagnosed with extensive-stage SCLC, in the 12 months covered by this report, the FDA approved expanding the use of both nivolumab and pembrolizumab to include treating patients who have extensive-stage SCLC that has progressed despite treatment with a platinum-based cytotoxic chemotherapeutic and at least one other cytotoxic chemotherapeutic. The August 2018 approval for nivolumab was based on results from a phase I/II clinical trial that showed that nivolumab treatment led to partial or complete tumor shrinkage in 12 percent of patients whose disease had progressed after previous treatments. For pembrolizumab, the June 2019 approval was based on results from two clinical trials, one a phase I and the other a phase II, that showed that pembrolizumab treatment led to partial or complete tumor shrinkage in 19 percent of patients whose disease had progressed after previous treatments. Most of the patients benefited from nivolumab or pembrolizumab treatment for 12 or more months.
From August 1, 2018 to July 31, 2019, the use of pembrolizumab was also expanded by the FDA to include the treatment of certain patients with an additional four types of cancer. In July 2019, the FDA approved this checkpoint inhibitor for treating patients who have recurrent, locally advanced, or metastatic squamous cell carcinoma of the esophagus that tests positive for PD-L1 protein and that has progressed despite treatment with at least one other therapeutic. The approval was based on results from two clinical trials, one a phase II and the other a phase III. In the phase III clinical trial, pembrolizumab significantly improved overall survival compared with cytotoxic chemotherapy. In the phase II clinical trial, pembrolizumab treatment led to partial or complete tumor shrinkage in 20 percent of patients, most of whom benefited from the treatment for 12 or more months.
In November 2018, pembrolizumab was approved by the FDA for treating patients who have hepatocellular carcinoma that has progressed despite treatment with the molecularly targeted therapeutic sorafenib, which has been the standard of care for patients with this disease for more than a decade (see Blocking the Blood Supply to Liver Cancer). The approval was based on results from a phase II clinical trial that showed that treatment with pembrolizumab led to partial or complete tumor shrinkage in 17 percent of patients (249). Most of these patients benefited from pembrolizumab treatment for 12 or more months.
A month later, in December 2018, the FDA approved pembrolizumab for treating certain patients with a rare, aggressive form of skin cancer called Merkel cell carcinoma. Specifically, it was approved for treating children and adults who have recurrent locally advanced or metastatic Merkel cell carcinoma after results of a phase II clinical trial showed that 54 percent of patients treated with pembrolizumab had complete or partial tumor shrinkage.
The other new FDA approval for pembrolizumab occurred in April 2019. The checkpoint inhibitor was approved for use in combination with the molecularly targeted therapeutic axitinib (Inlyta) for the initial treatment of patients who have advanced renal cell carcinoma, which is the most common type of kidney cancer. The combination of pembrolizumab and axitinib was approved after results from a phase III clinical trial showed that the combination significantly improved overall survival rates compared with sunitinib (Sutent), which is one of the most commonly used initial treatments for patients newly diagnosed with advanced renal cell carcinoma (250).
In May 2019, the FDA approved a second checkpoint inhibitor, avelumab, for use in combination with axitinib for the initial treatment of patients who have advanced renal cell carcinoma. The approval was based on results from a phase III clinical trial that showed that treatment with the combination of avelumab and axitinib significantly increased the time to disease progression compared with sunitinib (251).
Checkpoint inhibitors have yielded extraordinary benefit for patients with a diverse array of cancer types, but they can have adverse effects. The adverse effects arising from treatment with checkpoint inhibitors relate to the fact that these immunotherapeutics work by releasing brakes on the immune system. In a significant proportion of patients, this activation of the immune system leads to immune-related adverse effects. These adverse effects can affect any organ in the body and range from rash and local inflammation that can be treated with steroids and/or by temporarily discontinuing the treatment, to more severe adverse effects like thyroiditis and diabetes that need lifelong treatment with thyroid medications and insulin, respectively (252). If we are to predict which patients are likely to have severe immune-related adverse effects following treatment with a checkpoint inhibitor and design treatments to combat these effects without compromising the anticancer efficacy of the checkpoint inhibitor, we must better understand why and how the severe immune-related adverse effects arise. This is an area of intensive research investigation (252-253).
Directing the Immune System to Cancer Cells
An immune cell must find a cancer cell before it can destroy it. Many immunotherapeutics that have been approved by the FDA for treating cancer work, at least in part, by helping immune cells find cancer cells (see Cell Lysis Mediators in Supplemental Table 2c). The most recent addition to this group of immunotherapeutics is mogamulizumab-kpkc (Poteligeo), which was approved by the FDA in August 2018 for treating certain patients with cutaneous T-cell lymphoma.
Cutaneous T-cell lymphomas are rare types of non-Hodgkin lymphoma that arise in immune cells called T cells. In these diseases, the cancerous T cells accumulate in the skin, resulting in an itchy, red rash. Mycosis fungoides and Sézary syndrome account for about two-thirds of the cases of cutaneous T-cell lymphomas diagnosed in the United States each year. In many cases, the cancerous T cells in patients with mycosis fungoides or Sézary syndrome have a protein called CCR4 on their surface. The newly approved immunotherapeutic mogamulizumab-kpkc targets CCR4. Once mogamulizumab-kpkc attaches to CCR4 on the surface of the cancerous T cells, it flags the cancer cells for destruction by immune cells.
The FDA approval of mogamulizumab-kpkc is for the treatment of adults who have mycosis fungoides or Sézary syndrome that has not responded to or has relapsed after treatment with at least one other treatment. The approval was based on results from a phase III clinical trial that showed that mogamulizumab-kpkc more than doubled the time to disease progression for patients compared with vorinostat (Zolinza), which is a standard treatment in this clinical setting (254).
One of the first immunotherapeutics to be approved by the FDA, rituximab (Rituxan), works, at least in part, by directing immune cells to cancer cells. Rituximab targets a protein called CD20, which is found on the surface of immune cells called B cells, both normal B cells and those that become cancerous. Since its first approval in 1997 for treating certain patients with a type of non-Hodgkin lymphoma called follicular lymphoma, which arises in B cells, it has been approved for treating several other types of non-Hodgkin lymphoma that arise in B cells and for treating CLL, which also arises in B cells.
Recently, researchers have been investigating whether combining newer anticancer therapeutics with rituximab can further improve outcomes for patients. In May 2019, the FDA approved a molecularly targeted therapeutic called lenalidomide for use in combination with rituximab for treating certain patients who have follicular lymphoma or another non-Hodgkin lymphoma that arises in B cells, marginal zone lymphoma. Specifically, the combination of lenalidomide and rituximab was approved for treating patients whose disease has progressed despite a previous treatment. The approval was based on results from two phase III clinical trials. In one of the trials, the time before disease progressed for patients with follicular lymphoma or marginal zone lymphoma that had progressed after initial treatment was almost three times longer among those who received lenalidomide and rituximab compared with those who received placebo and rituximab (255).

{kind=link}